Ants, Bees, Genomes & Evolution @ Queen Mary University London
Evolution of a Young Social Supergene in Fire Ants
June 10, 2022
Talk at College de France Supergene symposium
In Paris, Tatiana Giraud and her team including Jay Paul and Ricardo Rodriguez de la Vega organised a superb symposium on the evolution supergenes and sex chromosomes. The speaker lineup was superb, the food was great and the discussions were extremely stimulating. Yannick also had the honor of presenting our recent work on the fire ant social supergenes, highlighting that:
-
the Sb supergene responsible for multiple-queen colonies evolved in one species and repeatedly introgressed into other species. Thus the benefits of the new social form are sufficient to outweigh the genetic incompatibilities of carrying a large piece of chromosome from another species.
-
the young Sb supergene for which recombination is suppressed is accumulating deleterious mutations, including new transposable elements and expansions of existing repetitive elements. These have led the Sb supergene to almost double in size compared to SB. This degenerative expansion is an early stage of supergene evolution, that likely occurs in many other systems but is difficult to detect in them.
-
when comparing gene expression in the fire ant supergene system, most highly significant differences in gene expression are due to the genetic architecture of the system, and gene-by-gene dosage compensation for the degeneration of Sb, and only few patterns have the signature of socially antagonistic selection ELIFE paper.
Scientists find new colony structure of fire ants evolved in one species before spreading to others
March 11, 2022
Scientists from Queen Mary University of London have discovered that a new form of ant society spread across species. They found that after the new form of society evolved in one species, a “social supergene” carrying the instruction-set for the new social form spread into other species. This spread occurred through hybridisation, i.e., breeding between ants of different species. This unlikely event provides an alternate way of life, making the ants more successful than if they only had the original social form.
Red fire ants originally had only colonies with one queen. The team previously discovered that about one million years ago, a new social form evolved where colonies could have dozens of queens. A particular version of a large section of chromosome, named the “social supergene”, includes the genetic information necessary to make workers accept more than one queen. The new research, published today in Nature Communications, analysed the entire genomes or instruction sets of 365 male fire ants to examine the evolution of the social supergene, and found that the same version of this chromosome is present in multiple fire ant species.

Transfer of large amounts of genetic information across species is rare because of genetic incompatibilities. However, in this case, the advantages of having multiple queens overrode the incompatibilities, and the genetic material repeatedly spread to other species from the one source species in which this new social form evolved. The multiple-queen social form has advantages in several situations. For example, a multiple-queen colony has more workers and thus can outcompete a colony with only one queen. Furthermore, if there is a flood, a colony with multiple queens is less likely to become queenless.
Dr Yannick Wurm, Reader in Evolutionary Genomics and Bioinformatics at Queen Mary University of London and a fellow of The Alan Turing Institute said: “This research reveals how evolutionary innovations can spread across species. It also shows how evolution works at the level of DNA and chromosomes.
“It was incredibly surprising to discover that other species could acquire a new form of social organisation through hybridisation. The supergene region that creates multi-queen colonies is a large piece of chromosome that contains hundreds of genes. The many parts of a genome evolve to work together in fine-tuned manners, thus suddenly having a mix with different versions of many genes from another species is complicated and quite rare.
“Instead of executing extra queens as they would in a single-queen colony, the new version of the supergene leads workers to accept multiple queens. Having studied the history of the supergene and new social form extensively, we next want to identify which genes or parts of the supergene region, lead to these changes in behaviour. This will also help fill more gaps in our understanding of evolutionary processes.”
Rodrigo Pracana, a lead author of the study, also at Queen Mary University of London added: “Our study shows how detailed analysis of large numbers of wild animals can provide surprising new insight on how evolution works.”
The team from Queen Mary were previously among the first in the world to apply large-scale DNA-sequencing approaches to wild insects – which enabled them to discover one of the first well-known supergenes.
Red fire ants are native to South America and infamous for their painful sting. One of these species is known in many other parts of the world, where it is aggressiveness and high population density have made it an invasive pest. Efforts at controlling the spread of this species have largely been unsuccessful, as indicated by its Latin name, Solenopsis invicta, meaning “the invincible”.
The research was supported by the Leibniz Institute for the Analysis of Biodiversity Change, with Dr. Eckart Stolle assisting as part of the team at Queen Mary before continuing this work at the Leibniz Institute.
Dr Yannick Wurm is a Reader in Evolutionary Genomics and Bioinformatics at the School of Biological and Behavioural Sciences. His lab runs between Queen Mary University of London and The Alan Turing Institute. The lab studies the lives of social insects including ants and bees, and how they evolve in the face of social and environmental challenges. For this, lab members combine behavioural experiments with cutting-edge molecular approaches including genomics and bioinformatics. Find out more about the Wurm Lab.
Further information
Research publication: ‘Recurring adaptive introgression of a supergene variant that determines social organisation’ Stolle et al. Nature Communications
New study pinpoints bumblebee genes that help them adapt to environmental changes
February 9, 2022
Researchers from Queen Mary University of London and Imperial College London have identified genes that could help bumblebees overcome environmental challenges such as climate change.
The study, published in the journal Molecular Biology & Evolution, looked at the genome sequences, or DNA blueprints, of bumblebees from the widespread European species Bombus terrestris to understand how this species had been adapting to recent changes in the environment.

The research team then used evolutionary data science approaches to identify which parts of these DNA blueprints had been replaced by newer versions over recent decades. They found signs of recent changes to the genome in areas known to be linked to the nervous system and wing development.
The researchers suggest that the observed genetic changes likely improved the bumblebees’ abilities to forage for food in response to changes in climate and within their rural environments.
Interestingly, the researchers also uncovered some unusual features of the bumblebee genome, including a region containing 53 genes that lacked the diversity found in the rest of the genome.
Bumblebees and other insects are important natural pollinators of crops and of wildflowers. Recent studies have documented international declines of bees and other pollinators, citing habitat loss, disease, pesticides, and climate change as contributing factors. However, some of these pollinators, such as Bombus terrestris have also been doing well despite changing environmental conditions.
Lead author of this study Dr Yannick Wurm, Reader in Evolutionary Genomics and Bioinformatics at Queen Mary University of London and fellow of The Alan Turing Institute, said: “We found signatures of recent adaption in genes throughout the bumblebee genome, including for genes involved in the nervous system and in wing development.
“Simultaneously analysing many genomes of the buff-tailed bumblebee sheds new light on the health of this species. This species is doing well, and we found that most of the genome harbours extensive genetic diversity and the ability to use it. These traits will support this species in continuing to adapt to the challenges it faces.”
Dr Colgan, the first author of the study, said: “In contrast to the high genetic diversity in most of the genome, we found a gene-rich region with the opposite pattern: extremely low diversity in bumblebees and in their relatives, the honeybees. We don’t fully know the evolutionary reasons for the pattern observed in this region of the genome, nor how it may impact the ability of the species to adapt.”
The findings provide important insights into the ability of a key pollinator to adapt and highlight the benefit of genomic approaches for understanding the genetic health of wild populations. The researchers suggest that this type of approach could help develop tools for safeguarding beneficial insects important for ecosystem stability, biodiversity maintenance and crop productivity.
“The UK hosts more than 1,500 beneficial pollinator species, including many species of bees,” added Dr Wurm. “Applying the genomic approach developed here to other pollinators can help identify those species most at risk and inform the development of custom-tailored conservation and mitigation strategies.”
Dr Yannick Wurm is a Reader in Evolutionary Genomics and Bioinformatics at the School of Biological and Behavioural Sciences. His lab runs between Queen Mary University of London and The Alan Turing Institute. The lab studies the lives of social insects including ants and bees, and how they are affected by social and environmental challenges. For this, lab members combine behavioural experiments with cutting-edge molecular approaches including genomics and bioinformatics. Find out more about the Wurm Lab.
UNIX/bioinformatics teaching cloud
November 1, 2020
Getting into big data science can be a big leap if you’re a biologist who is new to the command-line.
We try to cut that down into a series of smaller, more manageable steps.
As part of that, we run a hands-on genome bioinformatics course that introduces students to UNIX, and covers topics from Illumina read cleaning to genome assembly, annotation, population genomics and genome-wide association mapping.
For obvious 2020 reasons, we needed to do this online in a manner that:
- has manageable costs but sufficient power for genomics analyses;
- is easy for students to access autonomously;
- provides students the flexibility to work when they want (timezones) from where they want;
- can be easily modified by us as needed.
- doesn’t require students to install complex software (Docker, Virtual box, linux subsystem…) which are difficult to troubleshoot.
We built it.
Process for students
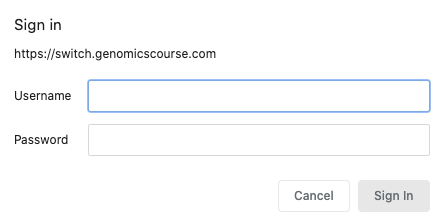
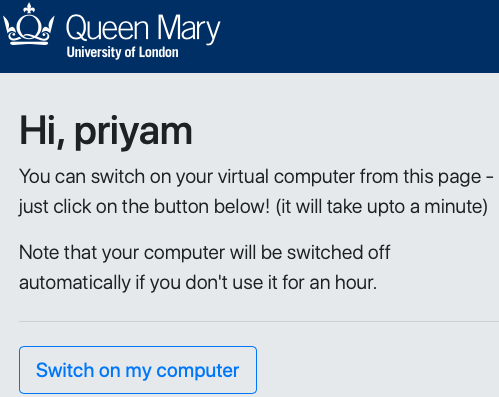
A student wanting to access their Linux machine must:
- connect to http://switch.genomicscourse.com (currently offline)
- enter their login/password
- click a button to switch on their virtual computer
- this creates their personal virtual computer and shows its IP address & hostname
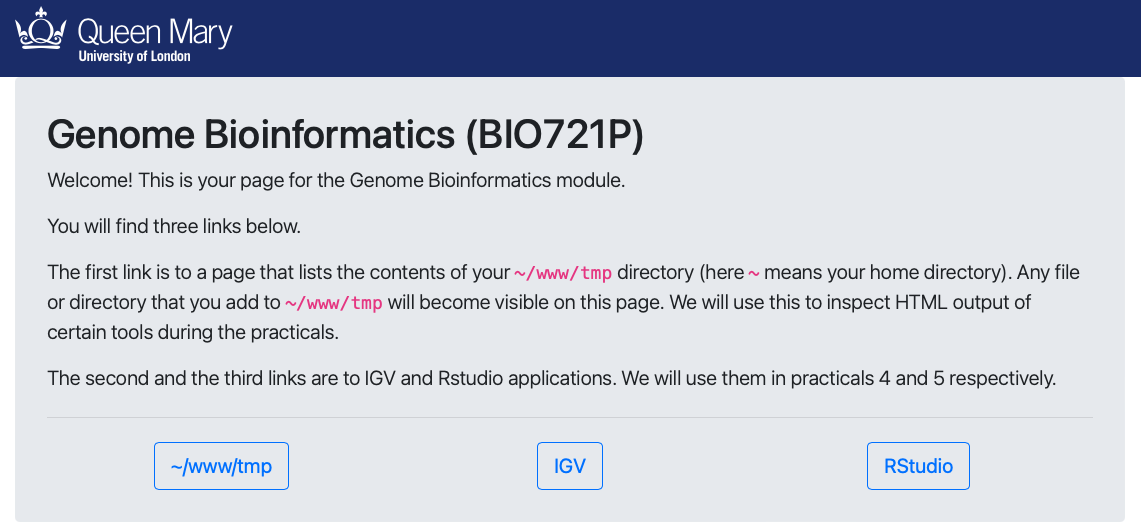
The student can connect to the computer by ssh, and download or visualise files in a web browser by putting them in a designated folder in their home directory (~/www).
If a student forgets to switch off their computer, this occurs automatically after 30 minutes of idle time.
For course administrators
This is great for us as organisers because:
- it avoids paying for cloud computing instances that are not being used.
- it allows us to give students more CPU and RAM.
- no physical rooms required - anyone can connect from any computer.
- ensures that all students use the same setup.
This uses Amazon EC2 infrastructure, and thus scales easily to any number of students, and can use computers with small or large amount of cpu power or ram.
Interested?
We can potentially deploy our solution for other courses. If you’re interested, get in touch.
Example screenshots
New study shows evolutionary breakdown of ‘social’ chromosome in ants
August 25, 2020
Scientists from Queen Mary University of London have found that harmful mutations accumulating in the fire ant social chromosome are causing its breakdown.
The chromosome, first discovered by researchers at the University in 2013, controls whether the fire ant colony has either one queen or multiple queens. Having these two different forms of social organisation means the species can adapt easily to different environments and has resulted in them becoming a highly invasive pest all over the world, living up to their Latin name Solenopsis invicta, meaning “the invincible”.

For the new study, published in eLife, the research team performed detailed analyses of the activity levels of all the genes within the social chromosome for the first time to understand how it works and its evolution. They found that damaging mutations are accumulating in one version of the social chromosomes, causing it to degenerate.
The findings also showed that most of the recent evolution of these chromosomes stems from attempts to compensate for these harmful mutations.
Overcoming evolutionary conflict
Natural selection is the main evolutionary mechanism that helps to optimise genes over generations but normally, it cannot simultaneously optimise genes for two different types of social organisation within one species.
To overcome this evolutionary conflict, social chromosomes group together genes adapted to each type of social form. The results of the new study show that this solution prevents the removal of harmful mutations from the genome and as a result, these mutations accumulate over time and begin to dominate the fate of the system.
The social chromosomes in fire ants are a rare example of a direct link between genes and social behaviour. They work in a similar way to the X and Y chromosomes in humans, which determine sex.
This discovery has wider ecological and medical implications because genomic structures similar to social and sex chromosomes can not only help species adapt to changing environments but also underpin diseases such as cancer.
Initial benefit has a long-term cost
Dr. Martínez-Ruiz, lead author of the study from Queen Mary University of London, said: “Our results show that the initial benefit of nature combining genes into a social chromosome has a cost. One million years later, most of the differences we see between social chromosomes are due to the accumulation of negative mutations.”
“We also see that the rest of the genome adapts very quickly in response to negative mutations,” added Dr. Wurm, Reader in Bioinformatics at Queen Mary and senior author of the study. “This is how evolution works, by adding patches to imperfect solutions, rather than by finding the most efficient solution.”
“Despite the degeneration of the social chromosomes, the fire ants are unlikely to lose them anytime soon. This would require another major chromosomal reshuffling – such events are rare and usually lethal,” Dr Wurm continues. “However, over long evolutionary timescales, anything is possible. Most of the 20,000 species of ants either have only single-queen colonies or only multiple-queen colonies. We are now trying to understand whether social chromosomes are required for changes in social organisation.”
The study builds on earlier research by the authors on the evolution of social chromosomes. They have previously identified differences in genes for chemical communication that may be responsible for perceiving queens, showed that one social chromosome has doubled in size, and that this social chromosome lacks genetic diversity.
More information
-
Research paper: ‘Genomic architecture and evolutionary antagonism drive allelic expression bias in the social supergene of red fire ants’ Carlos Martinez-Ruiz, Rodrigo Pracana, Eckart Stolle, Carolina I. Paris, Richard A. Nichols, and Yannick Wurm, 2020. eLife, 9, p.e55862.
-
The Wurm lab studies the lives of social insects including ants and bees, and how they are affected by social and environmental challenges. For this, lab members combine behavioural experiments with cutting-edge molecular approaches including genomics and bioinformatics. More information at wurmlab.com.
Press release: Degeneration of Gene Expression on Social Supergene
August 25, 2020
Scientists from Queen Mary University of London have found that harmful mutations accumulating in the fire ant social chromosome are causing its breakdown.
The chromosome, first discovered by researchers at the University in 2013, controls whether the fire ant colony has either one queen or multiple queens. Having these two different forms of social organisation means the species can adapt easily to different environments and has resulted in them becoming a highly invasive pest all over the world, living up to their Latin name Solenopsis invicta, meaning “the invincible”.
For the new study, published in eLife, the research team performed detailed analyses of the activity levels of all the genes within the social chromosome for the first time to understand how it works and its evolution. They found that damaging mutations are accumulating in one version of the social chromosomes, causing it to degenerate. The findings also showed that most of the recent evolution of these chromosomes stems from attempts to compensate for these harmful mutations. Natural selection is the main evolutionary mechanism that helps to optimise genes over generations but normally, it cannot simultaneously optimise genes for two different types of social organisation within one species.
To overcome this evolutionary conflict, social chromosomes group together genes adapted to each type of social form. The results of the new study show that this solution prevents the removal of harmful mutations from the genome and as a result, these mutations accumulate over time and begin to dominate the fate of the system.
The social chromosomes in fire ants are a rare example of a direct link between genes and social behaviour. They work in a similar way to the X and Y chromosomes in humans, which determine sex. This discovery has wider ecological and medical implications because genomic structures similar to social and sex chromosomes can not only help species adapt to changing environments but also underpin diseases such as cancer.
Dr. Martínez-Ruiz, lead author of the study from Queen Mary University of London, said: “Our results show that the initial benefit of nature combining genes into a social chromosome has a cost. One million years later, most of the differences we see between social chromosomes are due to the accumulation of negative mutations.”
“We also see that the rest of the genome adapts very quickly in response to negative mutations,” added Dr. Wurm, Reader in Bioinformatics at Queen Mary and senior author of the study. “This is how evolution works, by adding patches to imperfect solutions, rather than by finding the most efficient solution.”
“Despite the degeneration of the social chromosomes, the fire ants are unlikely to lose them anytime soon. This would require another major chromosomal reshuffling - such events are rare and usually lethal,” Dr Wurm continues. “However, over long evolutionary timescales, anything is possible. Most of the 20,000 species of ants either have only single-queen colonies or only multiple-queen colonies. We are now trying to understand whether social chromosomes are required for changes in social organisation.”
The study builds on earlier research by the authors on the evolution of social chromosomes. They have previously identified differences in genes for chemical communication that may be responsible for perceiving queens, showed that one social chromosome supergene variant has doubled in size, and that this social chromosome supergene lacks genetic diversity.
Research paper: ‘Genomic architecture and evolutionary antagonism drive allelic expression bias in the social supergene of red fire ants’ Carlos Martinez-Ruiz, Rodrigo Pracana, Eckart Stolle, Carolina I. Paris, Richard A. Nichols, and Yannick Wurm, 2020. eLife, 9, p.e55862. Available at https://doi.org/10.7554/eLife.55862.
Workers of the red fire ant Solenopsis invicta on an llumina Miseq sequencing chip used to analyse the genes in their social chromosome (credit: E Favreau & Y Wurm).
Getting a good internet connection from out in the boonies
July 24, 2020
Ok so Covid made Yannick et al head for the woods. But what about internet access?
It took some time but I figured it out. It is now fast and reliable. Low ping. Fast download. Fast upload. Amazing.
I suspect others in remote places could benefit from what I learnt. Two things were needed.

A high performance 4G/LTE router
The MikroTik Chateau is incredible.
Put a SIM card inside, and its already much faster than:
- a phone,
- a mifi hotspot,
- or a “simple” 90 euro 4G/LTE router like the Dlink DWR 921.
But plug in a dual antenna and it’s crazy fast.
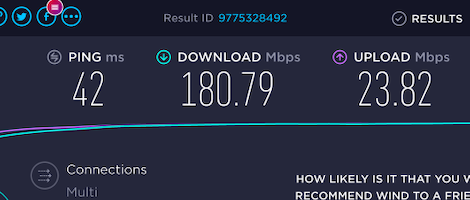
FYI, those 180 Mbps are despite having only 3 bars of reception.
It turns out that this router is Category 12 LTE, which means that it connects to the cell tower 3 times. So you get more combined bandwidth, and more resilience to interruptions - say if one of the cell towers were to become overloaded or fail.
MikroTik design and build these in Latvia. Most of their stuff is geared towards professionals. So the user interface offers immense flexibility - but is not easy to use. And it didn’t just “plug and play”.
FWIW the Microtik Chateau is ~200 GBP in UK. If out of stock, or you want something more user friendly, the Netgear Orbi is super-fast, or TP-link Archer MR600 is cheap but slower (Cat 6). All of these have antennas built in - so if signal is strong enough, no external antenna is needed.
A Yagi MIMO directional antenna on the roof
This had to be pointed at the nearest 4G phone tower - which I located using this handy map.
This made a huge improvement in cell reception.
(This type of pair of directional antennas is ~100 GBP). If you want to avoid the hassle of precise pointing, at the cost of lower sensitivity, get an omni-directional antenna that can just be stuck to the wall or window.

MikroTik Chateau configuration
Every small thing you could want to imagine can be tuned on this router - and bazillions of things I am not even close to imagining (!).
However, it didn’t work right away. I had to specifically:
1. Set the APN
Following MikroTik’s help documents, I did this in the Terminal interface:
/interface lte apn add apn=internet.it use-network-apn=no
/interface lte set lte1 apn-profiles=internet.it
2. In the Quick interface
- Specifically tell it to use both antennas
- Rename network, add password
- Update the OS to the latest development version
3. In the WebFig interface
- Tell it to accept incoming SMS
Getting internet while traveling
On the road (train, hotel rooms…), tethering to the iPhone is sometimes ok… but throughput is really much better with a dedicated device.
I stick a SIM card into a Netgear Aircard 790. Its wonderful and works right away; their newer NightHawk M2 is likely even better. If signal is weak, I take a portable mini LTE-antenna that can just be plugged into the aircard or nighthawk and stick that to the window. Obviously, if signal is very weak, a bigger antenna setup (like above) is needed…
Research Highlight: Degenerative expansion of the fire ant supergene
April 28, 2020
Non-recombining variant of a young supergene is larger than the normally-recombining variant
You know how a Y chromosome is usually smaller than an X chromosome, and it contains fewer genes?
We tested whether this pattern holds for the fire ant social chromosomes: is the b (which is similar to Y) smaller than the B (which is similar to X)?
Surprisingly, the opposite is true: the non-recombining b is almost twice as big as the B.

This is due to a process called “degenerative expansion”. We found the first evidence of it in an animal (there was a previous report in Papaya).
So why is the b variant of the fire ant supergene expanding?
It turns out that immediately after recombination is suppressed, massive loss of genes is too costly.
Instead, degeneration of a non-recombining region occurs in 3 steps:
- Accumulation of “mildly” deleterious mutations (e.g., repeats) ➜ Slow degradation of functional elements ➜ Degenerative expansion
- Compensatory mutations in the rest of the genome are selected for (e.g., dosage compensation, gene relocation)
- Cost of losing genes is lower ➜ Bigger chunks of the genome can now be lost
Why is it rare to observe the growth of a non-recombining region?
In most sex chromosome and other supergene systems, we have only seen the end of the 3rd phase. The systems studied are typically millions to hundreds of millions years old.
However, in the fire ant social supergene system we’ve been able to observe the first phase. This is probably for four reasons: the fire ant supergene is young, we used extra-long molecule sequencing, and ant males are haploid, making it easy to detect differences between haplotypes, and also creating strong purifying selection against gene loss.
For more details, check Eckart & Roddy’s paper: Degenerative expansion of a young supergene. Molecular Biology and Evolution 2019.
Succesful PhD & MSc completions & new members
April 25, 2020
Long time no update (!)
Party Time!
Huge congratulations for the labs three new 2019 PhD graduates:
- Emeline Favreau, now Postdoc with Seirian Sumner at UCL’s Biodiversity Research Centre;
- Carloz Martinez Ruiz, now postdoc at UCL’s Department of Oncolony;
- Leandro Santiago, now postdoc at the Wolfson Institute of Preventive Medicine.
Similarly, big congrats to the four MSc students we hosted in 2019: Valentine Patterson and Richard Burns are now pursuing PhDs (respectively in Mainz and at Kings College). Iwo Pieniak and Catherine Okuboyejo are now data engineers.
All will be sorely missed.
Additional congrats to the lab’s former postdocs Joe Colgan and Eckart Stolle, who have or are both moved to new junior group leader positions in Germany.
Hellos
Simultaneously we have welcomed a bunch of happy faces: Gabriel Hernandez-Gomez, Alicja Witwicka and Guy Mercer as new PhD students, and Anindita Brahma as a new Marie Sklodowska-Curie fellow. We look forward to working together!
Opportunities
As detailed on our phd and postdoc opportunities page, we are always eager to work with bright & motivated people, regardless what your background. Don’t hesitate to get in touch.
We will soon have an opening for a BBSRC-funded postdoctoral position looking at the molecular mechanisms underpinning the ability of pollinators to respond to environmental challenges. Stay tuned.
Improved regulation needed as pesticides found to affect genes in bees
March 7, 2019
Scientists are urging for improved regulation on pesticides after finding that they affect genes in bumblebees, according to research led by Queen Mary University of London in collaboration with Imperial College London.
For the first time, researchers applied a biomedically inspired approach to examine potential changes in the 12,000 genes that make up bumblebee workers and queens after pesticide exposure.

The study, published in Molecular Ecology, shows that genes which may be involved in a broad range of biological processes are affected.
They also found that queens and workers respond differently to pesticide exposure and that one pesticide they tested had much stronger effects than the other did.
Other recent studies, including previous work by the authors, have revealed that exposure even to low doses of these neurotoxic pesticides is detrimental to colony function and survival as it impairs bee behaviours including the ability to obtain pollen and nectar from flowers and the ability to locate their nests.
This new approach provides high-resolution information about what is happening at a molecular level inside the bodies of the bumblebees.
Some of these changes in gene activity may represent the mechanisms that link intoxification to impaired behaviour.
A major risk
Lead author of the study Dr Yannick Wurm, from Queen Mary’s School of Biological and Chemical Sciences, said: “Governments had approved what they thought were ‘safe’ levels but pesticides intoxicate many pollinators, reducing their dexterity and cognition and ultimately survival. This is a major risk because pollinators are declining worldwide yet are essential for maintaining the stability of the ecosystem and for pollinating crops.
“While newer pesticide evaluation aims to consider the impact on behaviour, our work demonstrates a highly sensitive approach that can dramatically improve how we evaluate the effects of pesticides.”
The researchers exposed colonies of bumblebees to either clothianidin or imidacloprid at field-realistic concentrations while controlling for factors including colony social environment and worker age.
They found clothianidin had much stronger effects than imidacloprid - both of which are in the category of ‘neonicotinoid’ pesticides and both of which are still used worldwide although they were banned in 2018 for outdoor use by the European Union.
For worker bumblebees, the activity levels of 55 genes were changed by exposure to clothianidin with 31 genes showing higher activity levels while the rest showed lower activity levels after exposure.
This could indicate that their bodies are reorienting resources to try to detoxify, which the researchers suspect is what some of the genes are doing. For other genes, the changes could represent the intermediate effects of intoxification that lead to affected behaviour.
The trend differed in queen bumblebees as 17 genes had changed activity levels, with 16 of the 17 having higher activity levels after exposure to the clothianidin pesticide.
Approach must be broadly applied
Dr Joe Colgan, first author of the study and also from Queen Mary University of London, said: “This shows that worker and queen bumblebees are differently wired and that the pesticides do not affect them in the same way. As workers and queens perform different but complementary activities essential for colony function, improving our understanding of how both types of colony member are affected by pesticides is vital for assessing the risks these chemicals pose.”
The researchers believe that the approach they have demonstrated must now be applied more broadly. This will provide detailed information on how pesticides differ in the effects they have on beneficial species, and why species may differ in their susceptibility.
Dr Colgan said: “We examined the effects of two pesticides on one species of bumblebee. But hundreds of pesticides are authorised, and their effects are likely to substantially differ across the 200,000 pollinating insect species which also include other bees, wasps, flies, moths, and butterflies.”
Dr Wurm added: “Our work demonstrates that the type of high-resolution molecular approach that has changed the way human diseases are researched and diagnosed, can also be applied to beneficial pollinators. This approach provides an unprecedented view of how bees are being affected by pesticides and works at large scale. It can fundamentally improve how we evaluate the toxicity of chemicals we put into nature.”
More information:
-
Research paper: ‘Caste- and pesticide-specific effects of neonicotinoid pesticide exposure on gene expression in bumblebees’. Thomas J. Colgan, Isabel K. Fletcher, Andres N. Arce, Richard J. Gill, Ana Ramos Rodrigues, Eckart Stolle, Lars Chittka and Yannick Wurm. Molecular Ecology.
-
Study Biology at Queen Mary